Parkinson's Disease
What is Parkinson's Disease?
Parkinson’s disease is a progressive neurological disorder which leads to shaking (tremor), stiffness, slowness of daily activities, and difficulty with walking, balance, and coordination.
Who can develop Parkinson’s disease?
- It is common in people over 60 years of age, but can occur in younger people as well.
- The symptoms usually begin gradually and get worse over time.
- About 5 in 1,000 people in their 60s and about 40 in 1,000 people in their 80s have PD. It affects men and women but is a little more common in men.
- Cases under 40 years of age consist of 10-15% of all cases and are known as young onset Parkinson’s disease.
What happens in PD?
- Parkinson’s disease affects the nerve cells in the substantia nigra of the brain that produce dopamine- a neurotransmitter messenger in the body that allows smooth co-ordinated movements.
- When a person has Parkinson’s disease, these dopamine producing cells start to degenerate and amount of dopamine produced in the brain decreases. Messages from the brain telling the body how and when to move are delivered abnormally, leaving a person incapable of initiating and controlling movements in a normal way.
- It is characterized by loss of approximately 60-80% of the dopamine producing neurons in substantia nigra before the motor symptoms appear, and a profound loss of dopamine in striatum.
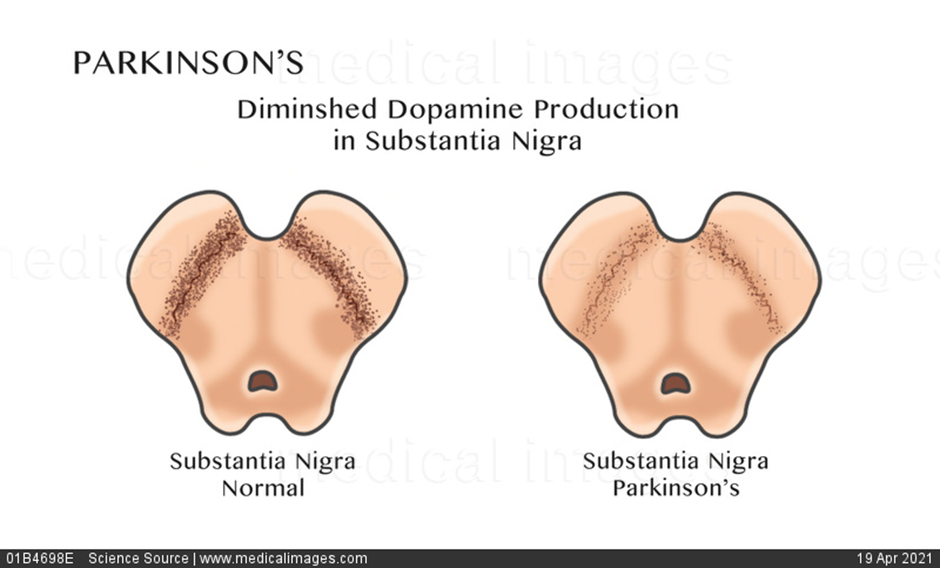
Parkinson's disease progression
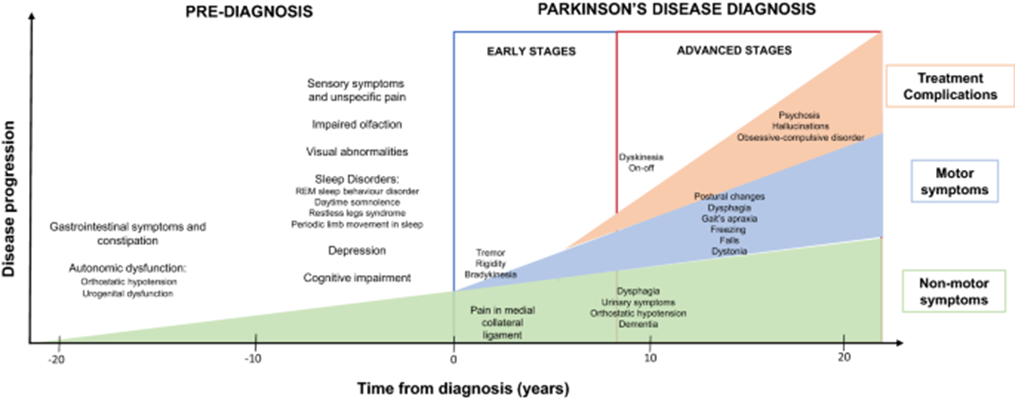
Chronology of clinical symptoms in Parkinson’s disease. Schematic representation of the diagnosis (even 10 to 20 years before the onset of the disease) and motor/non-motor symptoms in early and advanced Parkinson’s disease, with clinical and other iatrogenic symptoms.
What are the symptoms of Parkinson’s disease?
The symptoms of PD can be broadly classified as:
- Motor symptoms: They usually start on one side of body & usually spread to the other side of body as the disease progresses. These are:
- Tremors at rest of hands, legs, and/or jaw
- Slowness of speed of movements (bradykinesia)
- Stiffness of limbs (rigidity)
- Changes in posture, walking or balance difficulties
- Reduced facial expression (hypomimia)
- Difficulty in writing
- Change in voice quality or slurred speech (hypophonia, dysarthria)
- Difficulty in swallowing food (Dysphagia)
- Non-motor symptoms of PD: They can occur earlier in the disease before motor symptoms emerge and may be present throughout the course of disease. These are:
- Loss of smell sensations (hyposmia or anosmia)
- Altered taste sensations
- Memory decline (Dementia)
- Neuropsychiatric disturbances (depression, anxiety, hallucinations and psychosis)
- Constipation
- Sleep disturbances (inability to initiate or maintain sleep, excessive daytime sleepiness)
- Restless legs syndrome (RLS), REM sleep behavior disorder (RBD)
- Urinary problems
- Orthostatic hypotension
- Pain
- Abnormal excessive sweating or salivation
- Problems with controlling impulse e.g. compulsive shopping, eating, gambling and hypersexuality
- Loss of smell sensations (hyposmia or anosmia)
What are the stages of Parkinson’s disease?
- Stage 1: One side of body is affected
- Stage 2: Both sides of body affected but balance remains intact
- Stage 2.5: Both sides of body affected with mild impairment of balance
- Stage 3: Bilateral disease with impaired balance but functioning intact.
- Stage 4: Moderate to severe bilateral disease; walking and standing difficult without help
- Stage 5: Wheelchair bound or bed-ridden
- Not everyone with PD will progress to stage 4 or 5.
- Parkinson’s disease affects everyone differently & a patient may or may not have all the possible symptoms.
- Adhering to medication treatment advised by your Parkinson’s disease and Movement disorders specialist is very essential.
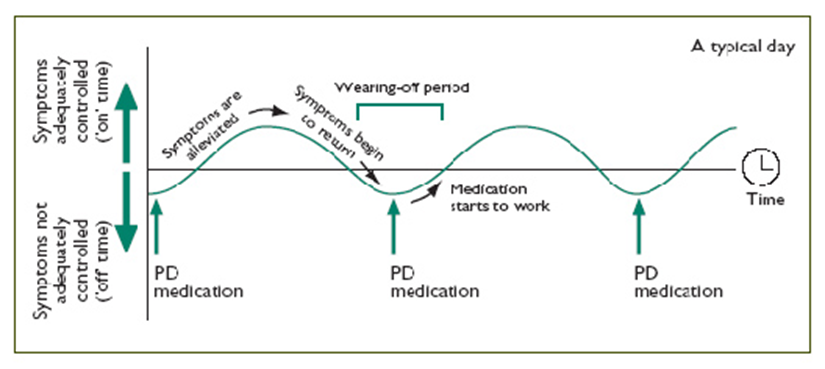
What is the “ON-OFF” phenomenon?
As PD progresses, patients may experience “ON-OFF” periods.
“OFF” periods are times when dopamine levels are low in the brain, and when the medicine is wearing off or not felt when it should be, presenting as motor or non-motor fluctuations. While “ON” periods are those when the patient doesn’t experience any motor (or non-motor) fluctuations and the effects of medications are good.
How can Parkinson’s disease be diagnosed?
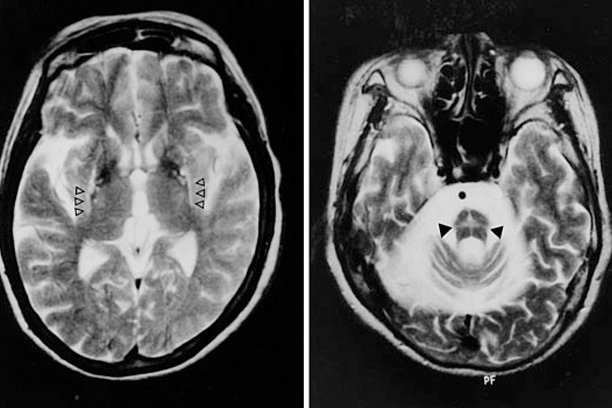
There is no single test that can diagnose your Parkinson’s disease. The diagnosis is mainly done by history and clinical examination of typical symptoms (described above) by your Parkinson’s disease and Movement Disorders Specialist. In the early stage of the disease, it may be difficult to differentiate between Parkinson’s disease and atypical parkinsonian syndromes due to mild and overlapping symptoms. As the symptoms gradually progress, the diagnosis becomes more obvious. Brain MRI often helps in differentiating PD from atypical parkinsonism.
What are the treatment options available for PD?
There are multiple medications available for Parkinson’s disease. Judicious use of these medications in a right combination can improve various symptoms. In selected advanced cases, Deep Brain Stimulation (DBS) surgery is an alternative form of treatment, which can improve medication refractory symptoms and quality of life.